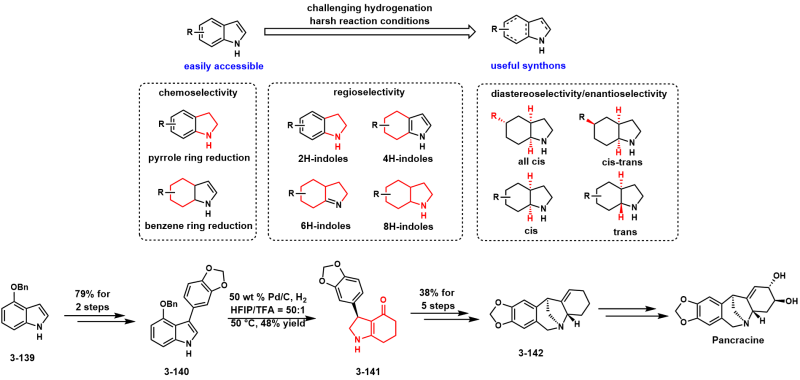
Bioinspired Formal Synthesis of Pancracine via Selective Hydrogenation of Indole Derivative
Shoule Han, Mingliang Lou Xiangbing Qi*
In drug development, sp3-hybridized carbons and heteroatoms are majorly observed in drug molecules1 and the saturated cores offer a higher possibility to access 3D spatial binding sites in target proteins than planar aromatic scaffolds. In particular, the chiral topology of the stereogenic centers can increase the druggability, including solubility, lipophilicity and stability, therefore highly associated with the ultimate clinical success rate. In terms of the atom and step economy, the chemo- and stereoselective hydrogenation of arenes and heteroarenes to synthesize three-dimensional saturated rings with multiple stereocenters has been proven powerful while challenging strategy due to the high activation energy required to break aromaticity.
Hydrogenases are a family of metalloenzymes that participate in the metabolism of hydrogen in organisms such as bacteria, archaea, and eukarya. Theoretically, hydrogenases activate the inert H-H bond (bond enthalpy: 436 kJ/mol) and promote the cleavage of dihydrogen into two protons accompanied by the generation of two electrons. Take the [Fe]H2ase hydrogenase (Figure 1a, right) as an example, H2 is cleaved heterolytically by placing H+ on the ligand O- group and hydride is placed on the Fe center (L=H-), which can then transfer hydride to substrates. Mimicking the hydrogenase enzymes, several organometallic catalysts have been devised to achieve effective performance in stereoselective hydrogenation of arenes and heteroarenes. For example, the ruthenium catalysts with a chiral diamine ligand promote the highly stereoselective reduction of quinolines,9–12 isoquinolines,13 quinolizidines,14 benzoxazines,15 and indoles.16,17 Hydrogenation of arenes has also been broadly utilized in the total synthesis of natural products,18–21 further demonstrating the power of this strategy.
DOI: 10.1055/a-2021-7944