Our lab is focused on the interface of synthetic methodology, natural product biomimetic synthesis and biologically valuable molecule discovery. We are encouraged by the rapid and efficient construction of molecular complexity from simple precursors in living organisms via enzymatic pathways. On the microcosmic molecular level, enzyme-catalyzed organic bond formation provided fundamental details of the molecular activation between catalyst and substrates through either non-bonded or covalent-bonded interactions. Inspired by the three-dimensional structure of the active binding residues of the enzyme pocket, we are interested in developing novel transformations for the stereoselective construction of chemical bonds for 3-D molecules that are otherwise tough to access. Privileged coordination of transition structure geometries and molecular recognition that control selectivity will be explored in detail by computational calculation and chemical kinetics. The utility of these stereoselective synthetic methodologies will be demonstrated by the synthesis of a variety of important biologically interesting compounds from biomass or simple feedstocks through an environmentally friendly cascade process that involves the formation of several chemical bonds and stereogenic centers simultaneously with excellent stereoselectivity. Structurally complex natural products are naturally produced in microorganisms through a series of elaborate biological pathways, so its biomimetic synthesis will be incorporated with dearomatization/molecular rearrangement reaction cascade in the lab. Besides the discovery of small molecule catalysis and natural product biomimetic synthesis, we also bear persistent enthusiasm for the pursuit of developing practical transformations to rapidly assemble novel bio-valuable molecules, which in turn serve as significant regulatory tools or pharmaceutical agents for a variety of biological pathways. In close collaboration with various elaborated labs at NIBS, we are devoted to stimulating synergistic cooperation among interdisciplinary fields to identify significant biological targets, interpret their mechanism of action and facilitate biomedical research for clinically valuable small molecule discovery. Based on high-throughput screening, computer-aided drug design and medicinal chemistry, drug development within the lab are highly focused on developing first-in-class therapies to address unmet medical needs, such as the inhibition of HBV infection, circadian clock or sleep regulation and anti-cancer via either small molecule induced protein degradation or molecular glue modulated protein-protein interaction.
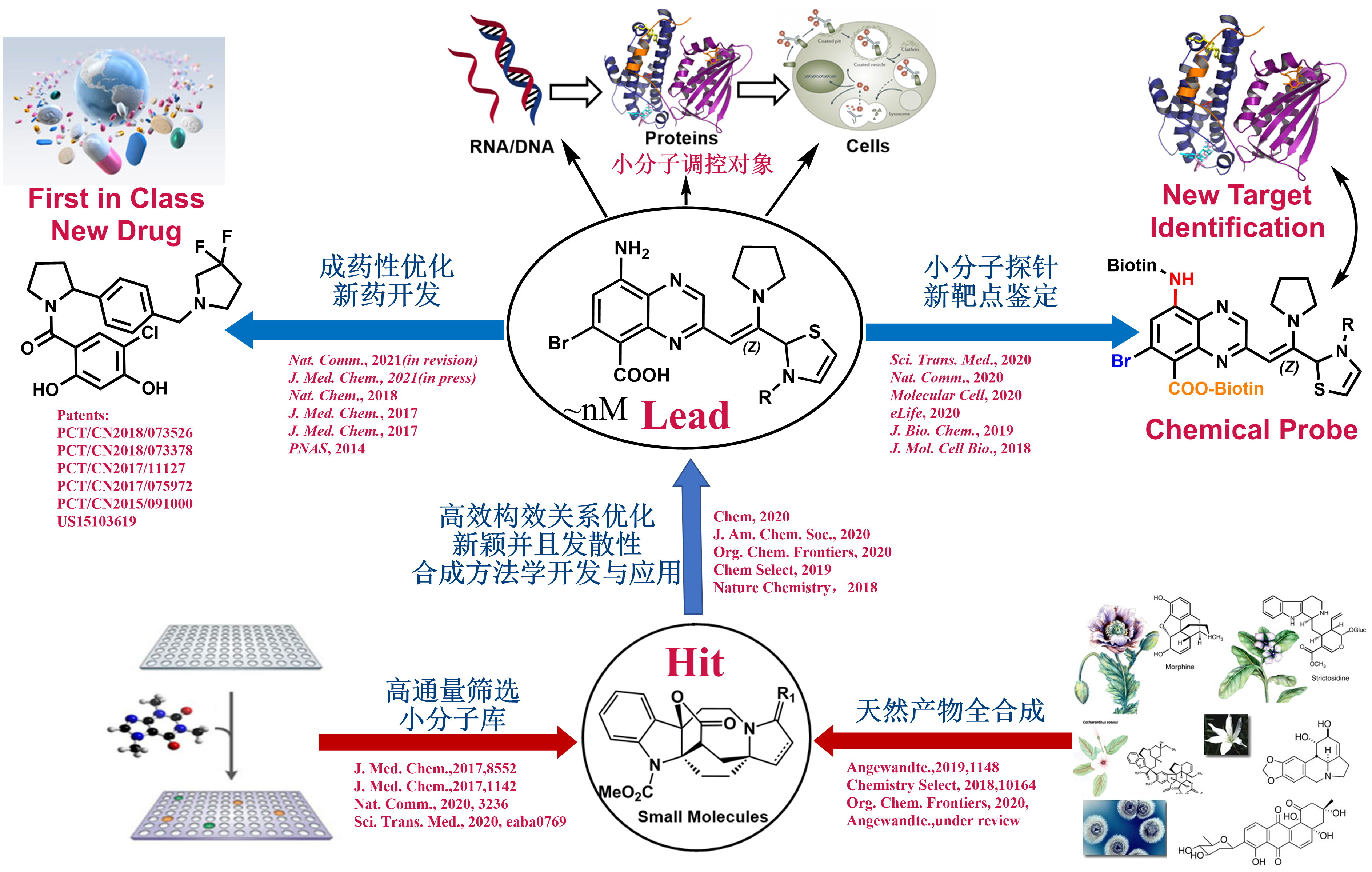
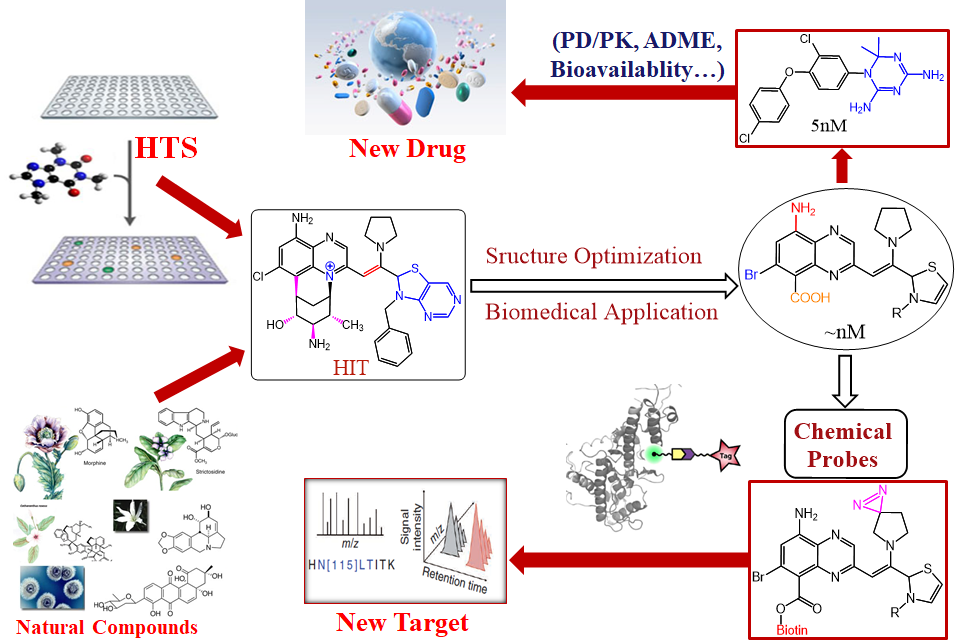
Drug Development Projects
The use of small molecules to probe systematic and disease-associated biological phenomena is an important aspect of our pursuits for exciting discoveries of biomaterials, chemical biology as well as pharmaceutical agents to solve serious health problems in a living system. Motivated by the urgent and valuable applications of small molecules in therapeutics and biomedical research, we are devoted to synthesizing small molecules in novel ways and examining their biological properties among a wide variety of biological areas conducted at NIBS. The outcomes of this biological evaluation and the performance in biological screening are the main guidance for further synthesis design. After a range of chemicals is screened against a particular drug target or disease model, and the qualified “hits” chemicals are concentrated and analyzed. Hit to lead development will be conducted to extend out the chemical library through “activity-oriented synthesis”. Lead optimization will be enforced with subtle and profound modifications to satisfy a variety of pharmaceutical rules before they become clinical candidates, including but not limited to solubility, oral bioavailability, cell membrane permeability, and many other known druggability features. In close collaboration with several biology labs at NIBS, we have developed one small molecule that regulates the circadian clock for 12 hours and identified the molecular target. We also discovered two novel types of molecular glue, one is promoting CDK12-DDB1 interaction to trigger Cyclin K degradation, another one inducing the phosphodiesterase 3A-schlafen 12 interaction to trigger the cancer cell apoptosis. We also identified the molecular target of an indole alkaloid, Nauclefine, and elucidated its mechanism of action in phosphodiesterase 3A-schlafen 12 dependent apoptosis. A highly potent and unique dimeric bile acid type of NTCP inhibitor for HBV infection and four novel kinase inhibitors towards BTK, LCK, IKKε, and PDK2 have been developed.
Chemical Development for the Circadian Rhythm Regulation
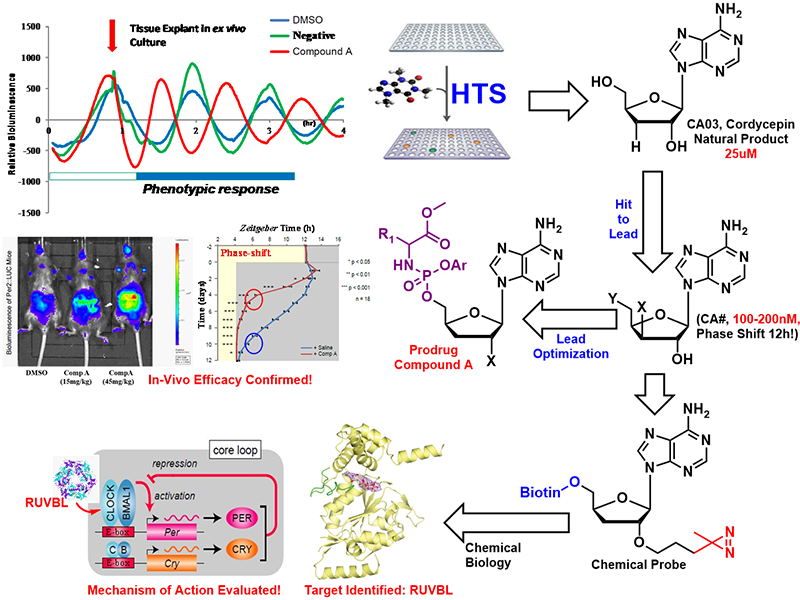
The circadian clock is tightly linked to human health. Clock malfunctions result in sleep disorders and lead to other diseases. The eukaryotic clock consists of a core oscillator that regulates its own transcriptional activity via a negative “Transcription-translation feedback loop” (TTFL), generating a roughly 24h rhythm in cells, tissues, and whole organisms. The TTFL model provides a dynamic view of how clock components work together to generate circadian oscillation.
In collaboration with Dr. Erquan Zhang lab in our institute, we found a nucleoside compound- 3′-deoxyadenosine (cordycepin)- that caused a robust and reliable “anti-phasic” phenotype, completely reversing (a 12h phase shift) the expression pattern of clock genes. Thorough SAR studies indicated that 3 additional nucleosides caused a phase-shift phenotype while nucleosides sharing the same sugar moieties with these hits but having different nucleobases did not shift the clock phase. Using simple peripheral injection treatment, we found that cordycepin could penetrate the blood-brain barrier and cause rapid entrainment of the circadian phase, facilitating reduced duration of recovery in a jet-lag model. We identified the interacting target of Cordycepin via a chemogenetic approach and chemoproteomics of Cordycepin-derived chemical probe. With the assistance from Dr. Huang lab, we also solved a crystal structure for RUVBL2 in complex with a physiological metabolite of cordycepin, and biochemical assays showed that cordycepin treatment caused disassembly of interaction between RUVBL2 and the core clock component BMAL1 .
The regulatory activity of this type of molecule was further optimized, leading to a new adenosine analog, CA04 with better DMPK properties and oral bioavailability. The prodrug strategy was executed, and the overall in-vivo efficacy was confirmed by the animal model. The ability of cordycepin/CA04 to penetrate the BBB, their rapid onset effect and their 12h phase reversal phenotype make them appear as the potential drug candidates to treat jet lag. Looking beyond the treatment of acute circadian clock disorders, one can envision that the ability to reliably and therapeutically entrain rhythmicity could help treat a broad swath of known clock-related chronic disorders as well. This work has been published in Science Translational Medicine (2020:eaba0769) and the related patent has been granted (WO2018133854A1).
Molecular Glue Development to Trigger Cyclin K Degradation
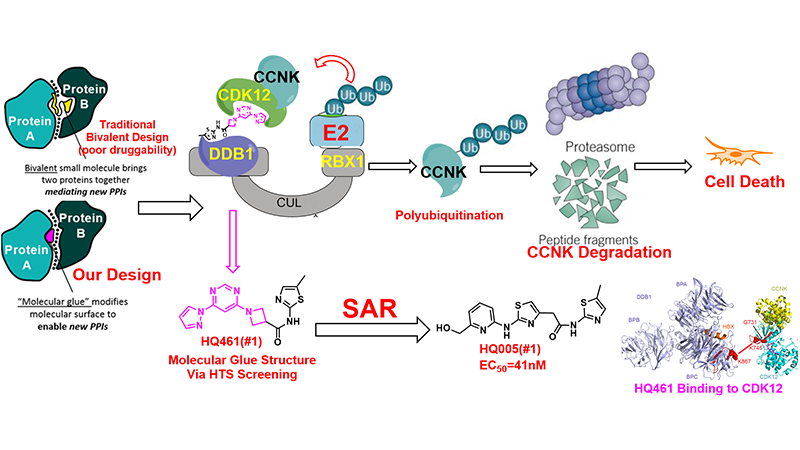
Inducing protein degradation through small molecules is one of the more promising strategies for treating cancer and suppressing viral infections. Targeted protein degradation strategies can achieve selective proteins degradation by binding to almost any site on the protein, especially for those that are traditionally considered non-druggable (~80%). The emergence of the proteolysis targeting chimera (PROTAC) technology makes it possible for any protein to be degraded via E3 ligation and ubiquitination. Specifically, the key of the technology is the design of a bifunctional molecule whose one end can bind to the target protein, the other end binds to E3 ligase. At present, the most popular PROTAC molecule is covalently linking the target binding drug and CRBN ligand pomalidomide or another small molecule ligand of VHL.
Molecular glues are small molecules that exert their biologic or therapeutic activities by inducing gain-of-function interactions between pairs of proteins. Molecular glue degraders, which mediate interactions between target proteins and components of the ubiquitin proteasome system to cause targeted protein degradation, hold great promise as a unique modality for therapeutic targeting of proteins that are currently intractable. In collaboration with Dr. Ting Han lab in our institute, we have developed a new molecular glue HQ461 by high-throughput screening and medicinal chemistry strategy. Using the unbiased loss-of-function and gain-of-function genetic screening followed by biochemical reconstitution, we show that HQ461 acts by promoting interaction between CDK12 and DDB1-CUL4-RBX1 E3 ubiquitin ligase, leading to polyubiquitination and proteasomal degradation of CDK12-interacting protein Cyclin K (CCNK). Degradation of CCNK mediated by HQ461 compromised CDK12 function, leading to reduced phosphorylation of a CDK12 substrate, downregulation of DNA damage response genes, and cell death. Structure-activity relationship analysis of HQ461 revealed the importance of a 5-methylthiazol-2-amine pharmacophore and resulted in an HQ461 derivate with improved potency (EC50, 41nM). Our studies reveal a new molecular glue that recruits its target protein directly to DDB1 and bypasses the requirement of a substrate-specific receptor, presenting a new strategy for targeted protein degradation. This work has been published in eLife (2020: articles/59994) and the related patent has been filed (PCT/CN2020/095482).
Identification of the Molecular Target of Nauclefine
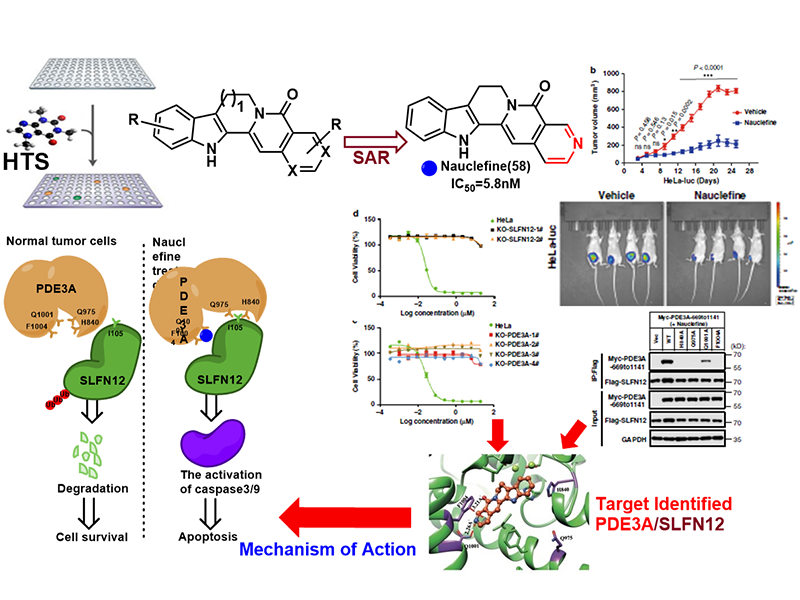
In collaboration with Dr. Xiaodong Wang lab in our institute, we identified that the nauclefine induces apoptosis of HeLa cells via a PDE3A-SLFN12 dependent cell death pathway. The direct binding of nauclefine with PDE3A was elucidated via chemogenetic and biochemistry approaches, and this binding induces the subsequent interaction of PDE3A and SLFN12. Nauclefine binds PDE3A but does not inhibit the phosphodiesterase activity of PDE3A, representing a new type of PDE3A modulator that initiates apoptosis without affecting its canonical function. In HeLa cells, the nauclefine-initiated PDE3A-SLFN12 interaction increases the protein stability of SLFN12, thereby promoting its apoptosis-inducing effects. Nauclefine was also shown in vivo to inhibit tumor xenograft growth in the PDE3A- and SLFN12-dependent manner, which illustrates PDE3A as a target for the development of novel anti-cancer therapeutics. This work has been published in Nature Communications, (2020, 11:3236).
BTK/LCK/IKKε Kinase Inhibitor
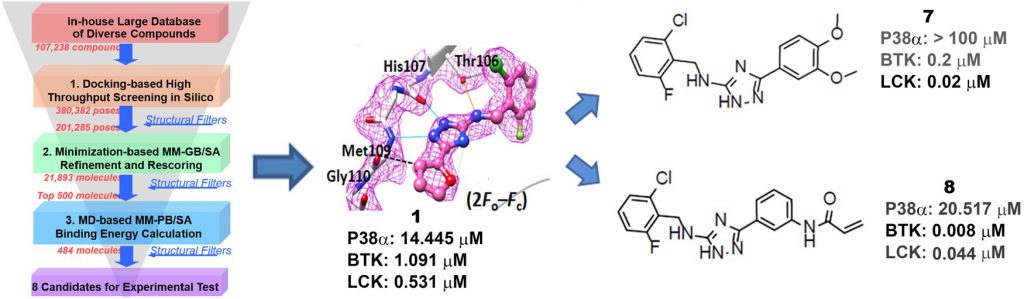
It is well established that the kinase gatekeeper residue plays a central role in determining the selectivity of kinase inhibitors. About 20% of human protein kinases have a small gatekeeper residue (e.g., glycine, alanine, cysteine, serine, or threonine) that directly affects the ability of drugs to access this typically hydrophobic interacting pocket. BTK and LCK are known to be important for the activation of lymphocytes. BTK is a member of the TEC subfamily that functions as a critical component in the B-cell antigen receptor (BCR) signaling pathway. LCK is an SRC subfamily protein with an essential function in proximal T cell antigen receptor (TCR) signal transduction. The p38α MAPK plays a vital role in inflammation by regulating the biosynthesis of tumor necrosis factor (TNF) α, interferon (IFN) γ, and other cytokines via transcriptional and post-transcriptional mechanisms in response to extracellular stimuli.
To explore novel kinase hinge-binding scaffolds, we collaborated with Dr. Niu Huang lab and carried out structure-based virtual screening against p38αMAPK as a model system. With the assistance of developed kinase-specific structural filters, we identified a novel lead compound that selectively inhibits a panel of kinases with threonine as the gatekeeper residue, including BTK and LCK. These kinases play important roles in lymphocyte activation, which encouraged us to design novel kinase inhibitors as drug candidates for ameliorating inflammatory diseases and cancers. Therefore, we chemically modified our substituted triazole-class lead compound to improve the binding affinity and selectivity via a “minimal decoration” strategy, which resulted in potent and selective kinase inhibitors against LCK (18 nM) and BTK (8 nM). The potent on-target inhibition effects of these compounds observed in cellular assays highlight the promise of these novel compounds as candidate drugs for the treatment of cancers and inflammatory diseases. Moreover, our work underscores both the power and flexibility of in silico approaches for drug discovery and represents a successful example of how a “minimal decoration” strategy can be used to turn an initial lead compound into attractive candidate drugs. This work has been published in J. Med. Chem (2017, 60 (20), 8552) and the related patent has been filed (WO2017152842).
Inhibitor-κB kinase ε (IKKε) was recently identified as a repressor of viral infections by modulating type I IFNs. More studies also showed that IKKε plays a crucial role in the regulation of NF-κB activity and inflammatory hyperalgesia in mice. IKKε modulates inflammatory nociceptive sensitivity by activation of NF-κB–dependent gene transcription and is useful as a therapeutic target in the treatment of inflammatory pain. In collaboration with Dr. Niu Huang lab, we identified a new scaffold of IKKε inhibitor by the structure-based approach. We further analyzed the structure of the hit and conducted the minimal SAR based on the binding motif and the computer-aided drug design and finally recognized the modification site of the small molecule. Supported by the chemical reactivity of the functional groups and potential optimization direction, we then designed unique combinatorial chemistry for the high-throughput reaction for SAR optimization. The binding affinity was rapidly optimized to about 200-fold more potent than the original hit and a variety of small molecules with low nM affinity and high selectivity were validated in a couple of weeks. The in-vivo activities were also confirmed and the lead optimization including better oral bioavailability is under development (unpublished).
PDK Kinase Inhibitor
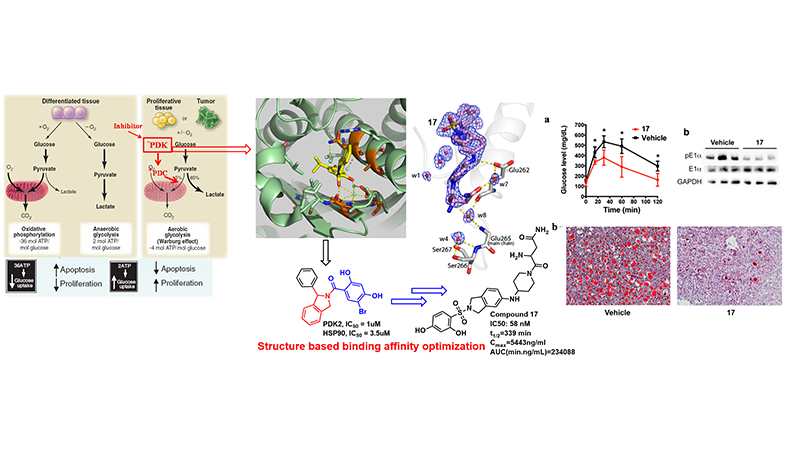
The macromolecular mitochondrial pyruvate dehydrogenase complex (PDC) catalyzes the oxidative decarboxylation of pyruvate to give rise to acetyl-CoA and is the gatekeeping enzyme linking glycolysis and the Krebs cycle. Because of its strategic location, the regulation of PDC activity is critical for glucose homeostasis and fuel preferences between glucose and fatty acids in the glucose-fatty acid cycle. Pyruvate dehydrogenase kinases 1-4 (PDK1-4) negatively control activity of the pyruvate dehydrogenase complex (PDC) and suppression of PDK activity impedes tumor growth, reduces glucose levels in diabetes, and improve bioenergetics in heart failure. Therefore, PDKs are potential therapeutic targets for these important human diseases. At present, there are no effective PDK inhibitors for preclinical or clinical studies targeting the PDKs.
We collaborated with Dr. David Chuang lab @UT southwestern and developed a novel PDK2 inhibitor targeting the ATP-binding pocket with high selectivity for PDK2 over the structurally related PDK1/3 and Hsp90. Hit compound was identified based on the comparison of crystal structures of the binding pockets in PDK and Hsp90. Hit to lead optimization resulted in a prominent feature of this type of inhibitor with preferential uptake and retention by the liver. This property confers significant augmentation of hepatic PDC activity, converting the liver from a normally gluconeogenic organ to a glucose oxidative machinery. Moreover, targeting PDK inhibitors to the liver may prevent extrahepatic toxicity and improve the efficacy of glucose-lowering therapeutics for the treatment of obesity and type 2 diabetes. This notion is further buttressed by the improved glucose tolerance with drastically reduced hepatic steatosis in 17-treated DIO mice. This work has been published in J. Med. Chem., (2017, 1142) and the related patent has been filed (WO2015089360).
Total Synthesis of Natural Products
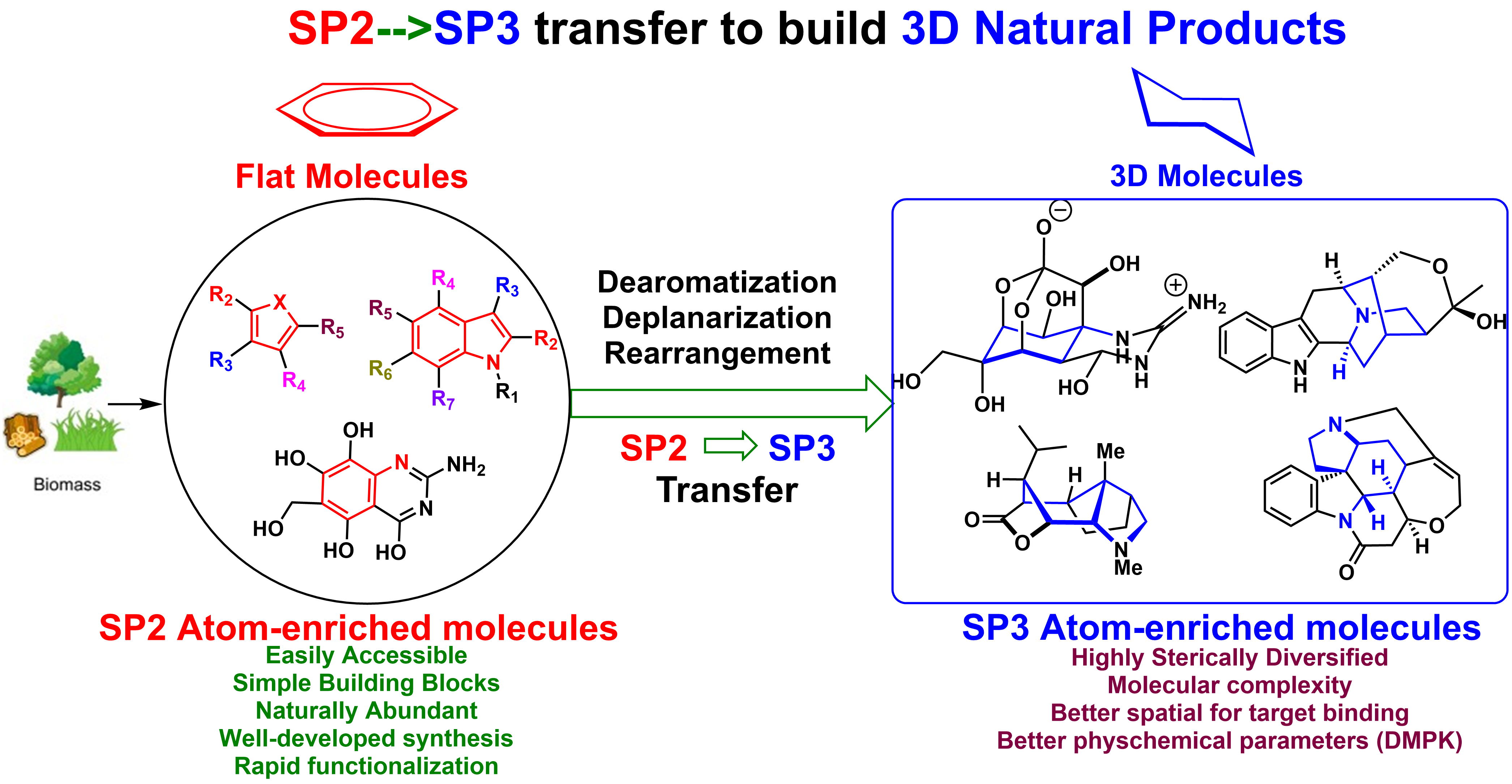
While the biological activity of small molecule and clinical success is highly correlated with greater complexity of the molecule, the increasing sp3 character of carbon may improve the complexity and therefore increase the opportunity to adjust the molecular shape by out-of-plane 3-D interaction of receptor/ligand that is not accessible for a flat aromatic ring, and thus improve potency and selectivity. These criteria have also been applied to the molecular complexity of natural product-like drug candidates by reducing the aromatic character of a molecule, which might further improve physical characteristics not only for better efficacy but also for better bioavailability.
Inspired by the rapid and efficient construction of molecular complexity from simple precursors in natural living systems, our lab is mainly focused on the logic of the total synthesis of sp3-atoms enriched natural product from biomass-derivatives or naturally abundant feedstocks. The utility of these renewable green building blocks has been demonstrated by the total synthesis of a variety of natural products through sp2 to sp3 carbon skeleton transfer strategy and environmentally friendly cascade process, which involves the formation of several chemical bonds and stereogenic centers simultaneously with excellent stereoselectivity. We developed a cascade reaction of chemoselective furan oxidation in combination with the controllable indole nucleophilic cyclization, which provides a rapid means for constructing a diverse set of structurally unrelated indole alkaloids and their unnatural analogues. Furan derivatives-based Aza-Achmatowicz rearrangement enabled the asymmetric total synthesis of (-)-alstofolinine A. Another sp2 to sp3 carbon skeleton transfer via indole hydrogenation strategy has been successfully effectuated and the resulting hydrogenated indole synthons have been applied into the total synthesis of architecturally complex natural products, such as lycorane-type alkaloids, strychnos, and akuammicine alkaloids. Besides the total synthesis, the mode of action study is highly investigated in the lab to reveal the role that natural products have and will continue to play in mapping important biochemical networks and identification of novel therapeutic targets.
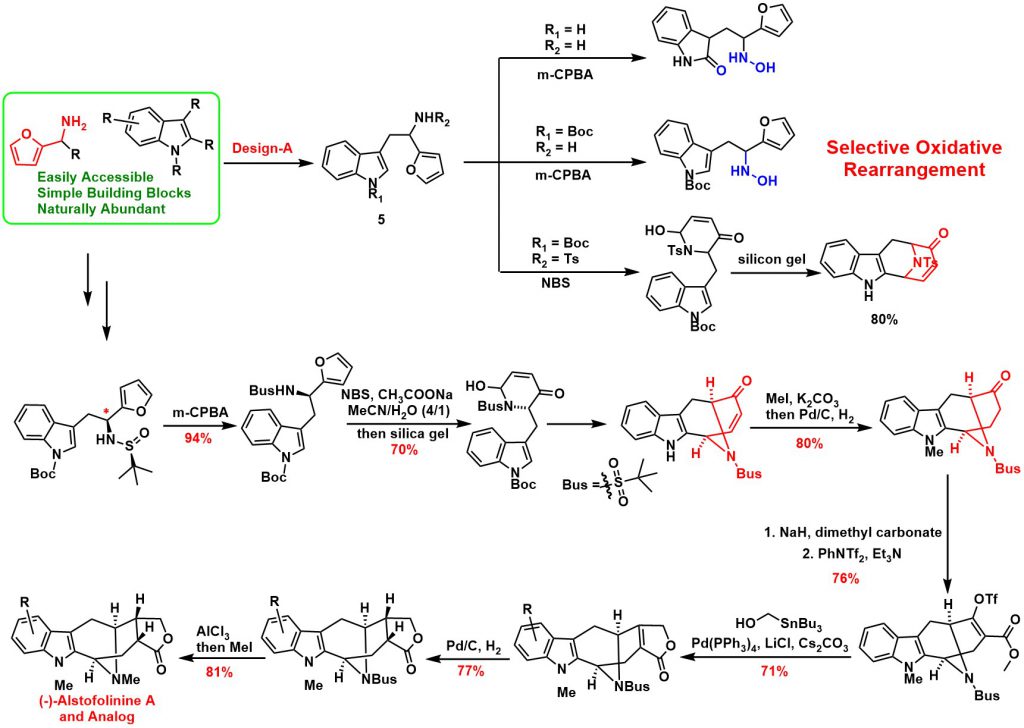
Developed a Novel Tandem Strategy of aza-Achmatowicz Rearrangement and Completed the Total Synthesis of (-)-Alstofolinine A
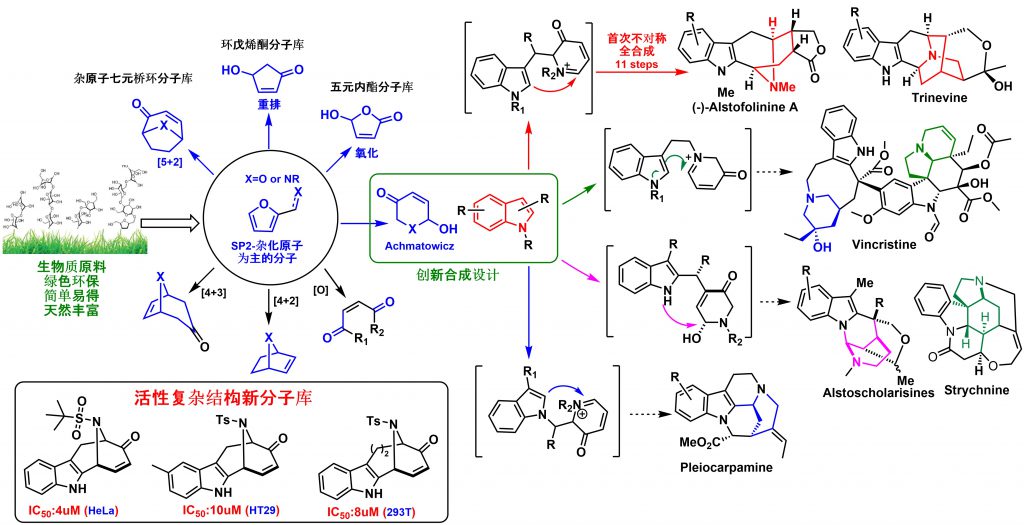
Furan-based compounds, which are predominately generated from biomass sources, are a family of important renewable green feedstocks. Furan derivatives have been broadly utilized in a variety of fields, including polymer matrix composites and resins, as well as more generally inorganic synthesis. Furans can also be used to synthesize complex molecules via selective oxidation/bromination and rearrangement, a process known as Achmatowicz rearrangement. Since 1971, Achmatowicz rearrangement has been incorporated into the total synthesis of hundreds of natural products. Aza-Achmatowicz rearrangement is an extension of the original Achmatowicz reaction to enable access to various nitrogenous ring systems embedded in many indole alkaloids. However, the study concerning aza-Achmatowicz rearrangement involving indole motifs has been less explored. Given the huge diversity of potential modifications on furan derivatives, we envisioned that aza-Achmatowicz rearrangement could potentially be adopted for a cascade reaction involving both furan and nucleophilic indole motifs. The strategic combination of aza-Achmatowicz rearrangement and subsequent indole nucleophilic cyclization could facilitate the construction of complex intermediates that would open the door for the total synthesis of a multitude of indole alkaloids.
Based on the above retrosynthetic strategies, we commenced our study with design-A. A reaction cascade of aza-Achmatowicz rearrangement followed by indole 2-position nucleophilic cyclization was explored and to our delight, the common indole-fused azabicyclo-[3.3.1]nonane core of the macroline family alkaloids could be generated successfully. The key to the success of the strategy relies on the careful manipulation of protecting groups and judicious selection of chemoselective furan oxidation conditions. The synthetic utility was further demonstrated on the asymmetric total synthesis of (-)-alstofolinine A. The target product was obtained with an overall yield of 7.3% in eleven steps. It is important to highlight that the chemoselective furan oxidation in combination with the controllable indole nucleophilic cyclization provides a rapid means for constructing a diverse set of structurally unrelated indole alkaloids and their unnatural analogues. This work has been published in Angew. Chem. Int. Ed. (2019, 4988).
Synthetic Methodology Projects
The use of small molecules to probe systematic and disease-associated biological phenomena is an important aspect of our research. Motivated by the urgent applications of small molecules in therapeutics and biomedical research, we are devoted to designing innovative strategies that assembly a wide variety of challenging complex molecules in a highly efficient manner for biomedical evaluation and novel function exploration.
Saturation of carbon or heteroatoms, especially sp3-hybridized and stereogenic atoms in pharmacophore allows the preparation of architecturally more complex molecules for the exploration of more diverse chemical space. While the biological activity of small molecule and clinical success is highly correlated with greater complexity of the molecule, the increasing sp3 character of carbon may improve the complexity and therefore increase the opportunity to adjust the molecular shape by out-of-plane 3-D interaction of receptor/ligand that is not accessible for a flat aromatic ring, and thus improve potency and selectivity. In our lab, we mainly focus on the synthetic logic and strategy for the activation, functionalization, and formation of sp3-carbon-carbone or sp3-carbon-heteroatom single bond. We, for the first time, developed the visible-light-induced single nickel-catalyzed C(SP3)–C(SP3), C(SP3)–C(SP2), and C(SP3)–C(SP) cross-coupling of alkylzirconocenes, which are easily prepared from unactivated alkenes either via steric-releasing “chain-walking” or Boron-directed “chain-walking”. High functional group tolerance and pharmacophore derivatization under mild conditions enable this method to be of practical significance. Furthermore, we have developed a highly enantioselective cross-coupling to build C(SP3)–C(SP3) single bond with chiral center asymmetrically. The mechanism studies supported a novel radical cross-coupling process and the synthetic potential of organozirconium will be extended within the lab.
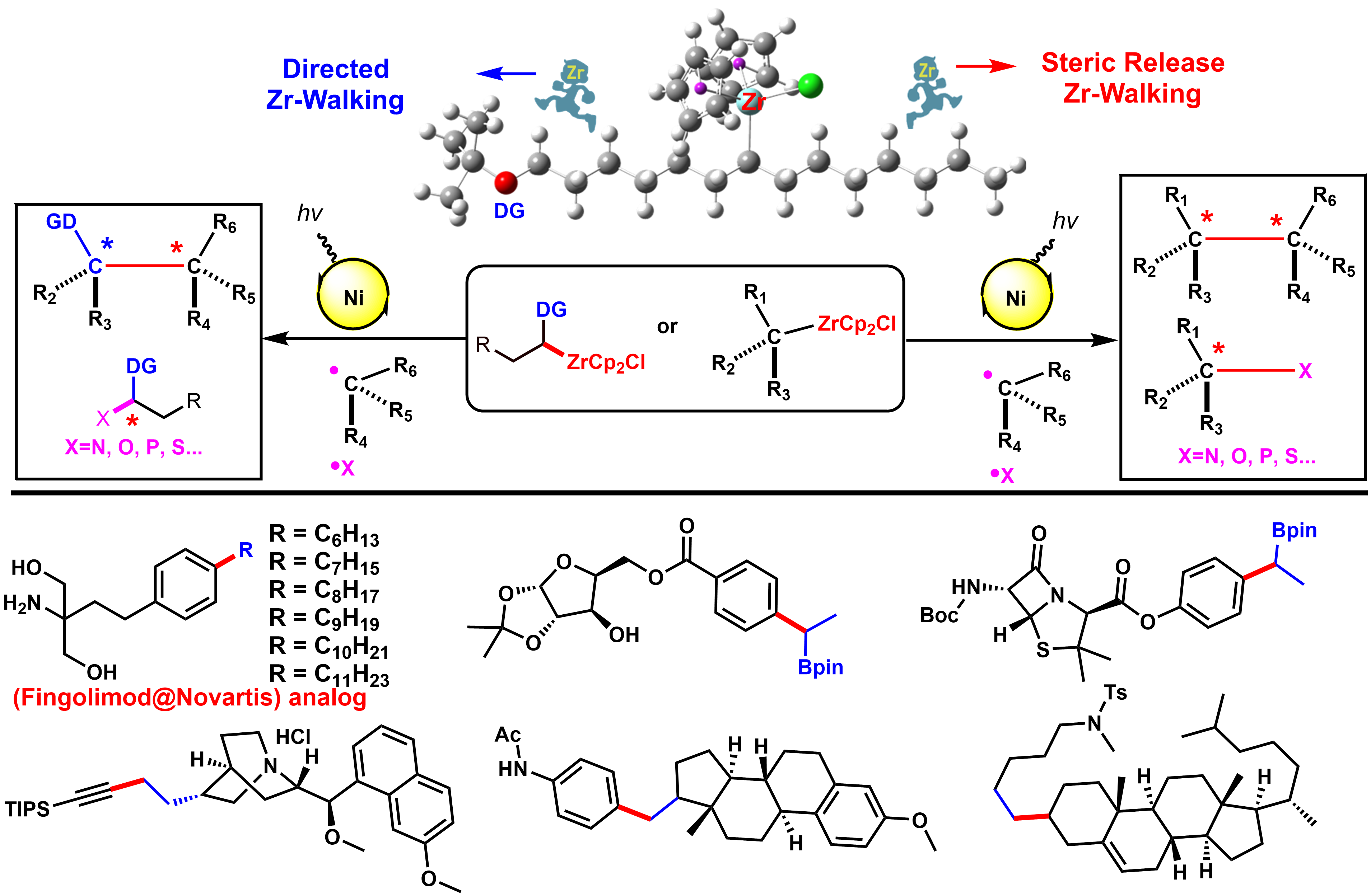
Visible-light-induced Single Nickel-catalyzed Cross-coupling of Alkyl-Zr
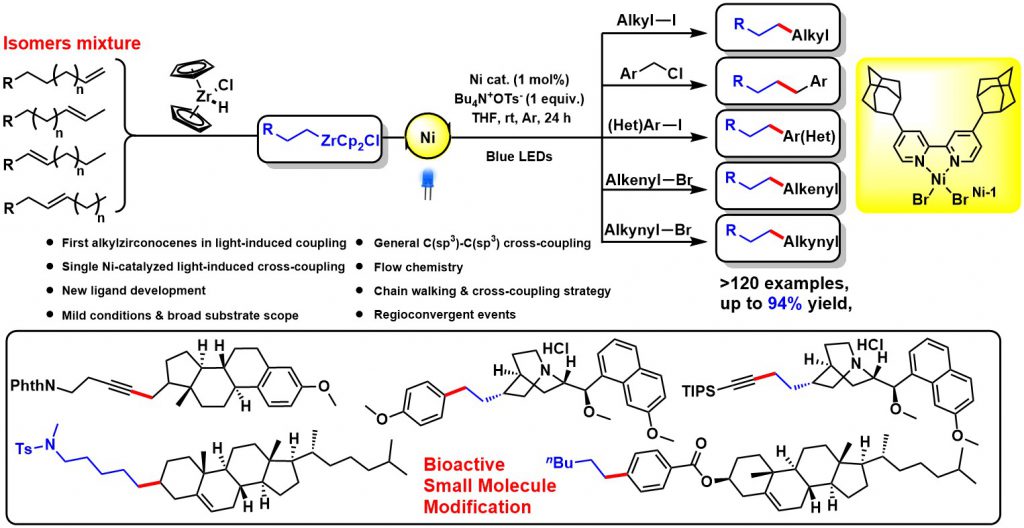
Transition-metal-catalyzed cross-coupling reactions between naturally abundant sp3-hybridized carbon centers facilitate access to diverse molecules with complex 3D structures, sp3-hybridized organometallic partners in cross-couplings are at present mainly limited to alkylmagnesiums, alkylzincs, and alkylborons. Organozirconocenes have emerged as very practical organometallic reagents, such as the homogeneous zirconium type Ziegler-Natta catalysts that are used in the synthesis of polymers of alkenes. Alkylzirconocene reagents, easily prepared from hydrozirconation of alkenes with bis(cyclopentadienyl)zirconium chloride hydride (Schwartz reagent), are capable of reacting with electrophiles to form various C-C(X) bonds. This represents one of the most attractive methods for value-added synthetic applications of unactivated alkenes. Compared with other alkylmetallic reagents, alkylzirconocenes offer several advantages, including mild generation conditions, excellent functional group tolerance, potential photoactivity, and remote functionalization through rapid “chain walking”. Therefore, we envisioned that alkylzirconocenes could be used as potential nucleophilic partners in cross-coupling reactions. Even though sp2-hybridized alkenyzirconocenes have gained increasing application in cross-coupling reactions, sp3-hybridized alkylzirconocenes have been far less explored-likely due to the lack of π-systems to stabilize the binding capability of zirconium to achieve suitable transmetalation.
We discovered a novel visible-light-induced single nickel-catalyzed C(sp3)-C(sp3), C(sp3)-C(sp2), and C(sp3)-C(sp) cross-coupling reactions using alkylzirconocenes. This method is mild and applicable for a large range of substrates including primary, secondary, tertiary alkyl, aryl, alkenyl, alkynyl halides, and a variety of alkenes. High functional group tolerance under mild conditions enables this method to be of practical significance. Employing a one-pot chain walking and cross-coupling relay strategy, a range of internal alkenes and an isomeric mixture of alkenes could be efficiently functionalized to give terminal coupled products. Synthetic applications including natural products and pharmacophore derivatization further demonstrated its advances of utilities and generalities. Mechanistic studies suggest a novel single nickel-catalyzed radical cross-coupling pathway, which represents the first visible-light-induced transformation of alkylzirconocenes. Given its mild conditions, scalability, and extremely general scope across a large range of organic halides, and considered alongside its excellent functional group compatibility, this strategy will serve as an innovative cross-coupling paradigm and will inspire researchers to revisit the synthetic potential of organozirconium. This work has been published in Chem (2020, 675).
Chemoselective Cross-coupling of Gem-Borazirconocene Alkanes
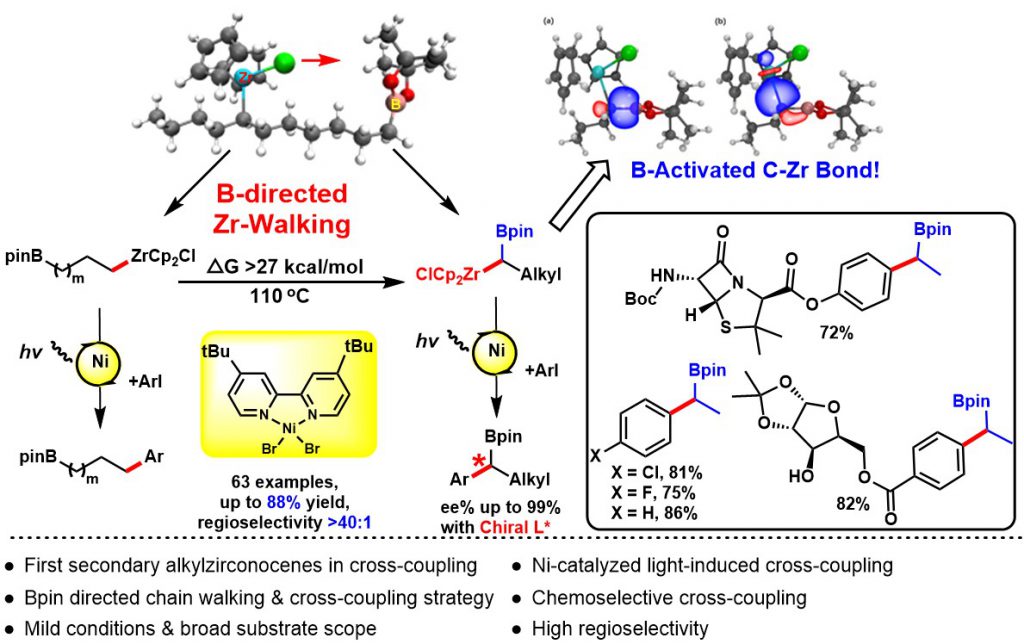
The development of remote functionalization methods is receiving growing interest, probably because such methods allow for the activation of challenging C-H and C-C bonds distant from the initiation position via a fascinating “chain walking” process. However, the manipulation of remote functional groups at different carbon centers represents a major challenge in synthetic chemistry. Hydrozirconation of internal alkenes produces primary alkylzirconocene complexes wherein facile Zr-H eliminations and reinsertions enable the Cp2ZrCl moiety to “chain walking” to the least sterically hindered terminal position of the alkyl chain. To the best of our knowledge, there is no general method to selectively direct this chain walking process to afford stable secondary alkylzirconocenes with predictable regioselectivity.
The selective cross-coupling of bimetallic alkyl nucleophiles is an attractive transformation to rapidly elaborate 3D structural complexity. Among reported bimetallic reagents gem-borazirconocene alkanes are recognized as particularly valuable synthons that possess great synthetic potential. This stable reagent is easily generated from hydrozirconation of various alkenyl boronic esters with bis(cyclopentadienyl)zirconium chloride hydride in high yield and with excellent functional group tolerance. The different reactivity of the carbon-zirconium bond vs the carbon-boron bond allows for the sequential functionalization of gem-borazirconocene alkanes via a variety of reactions such as cross-coupling, nucleophilic substitution, or nucleophilic addition. Organozirconium compounds are much more reactive towards electrophiles than organoboranes, which allows for the chemoselective conversion of organozirconium reagents to afford versatile organic boron intermediates. Despite the multiple advantages offered by these reagents, they have received substantially less attention for cross-couplings. While cross-coupling reaction of primary alkylzirconocene reagents has been reported by our group, to date, no general cross-coupling reaction has been developed for secondary alkylzirconocene including gem-borazirconocene alkanes, likely owing to the lack of available π-systems to stabilize the binding capability of zirconium to achieve suitable transmetalation. Furthermore, considerable steric hindrance around the carbon-zirconium bond further decreases its nucleophilicity.
We for the first time developed a Bpin group-directed “chain walking” process for alkylzirconocenes, selectively generating diverse gem-borazirconocene alkanes. We then further discovered a visible-light-induced single nickel-catalyzed chemoselective cross-coupling reaction of gem-borazirconocene alkanes with aryl halides, affording a wide range of alkylborane derivatives. Furthermore, by selectively harnessing the directing effect of the Bpin group, we have demonstrated that internal alkenes bearing a terminal Bpin can be converted into the desired coupled products with high regioselectivity. The utility of our method is also illustrated by the transformation of the Bpin intermediates, which, as well-established high-value building blocks, can be employed for lithiation-borylation, Zweifel olefinations, as well as the cross-coupling reaction that enable the rapid and efficient formation of sp3 carbon-carbon bonds. Additionally, we systematically investigated the Bpin-directed chain walking process underlying the regioselectivity of alkylzirconocenes, thus uncovering the mechanism of the remote functionalization of internal olefins achieved with our method. Finally, DFT calculations indicate that the high regioselectivity of this reaction originates from the directing effect of the Bpin group. This work has been published in Journal of the American Chemical Society (2020, 11506).
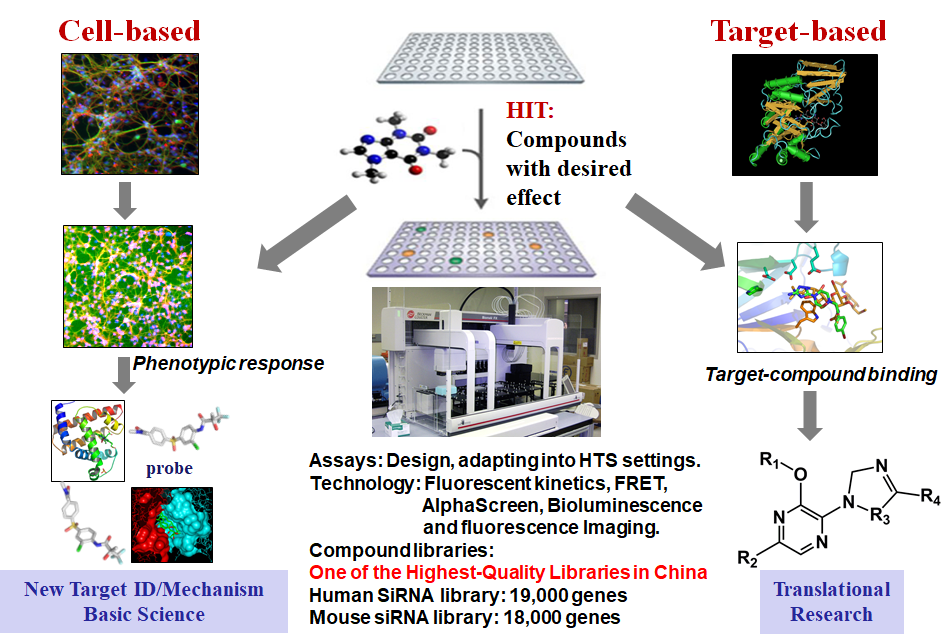
High-Throughput Screening
HTS is a drug-discovery process widely used in pharmaceutical companies and biomedical research institutes. Using robotics, data processing, and control software, liquid handling devices, and sensitive detectors, HTS conduct extremely scalable assay to test the biological or biochemical activity of a large number of small molecules for discovering active agents for receptors, enzymes, ion-channels or other pharmacological targets in the molecular and cellular level of biomolecular pathway. Typically, HTS assays are performed in microtiter plates with a 96 or 384 well format. HTS is one of the main facilities in our lab to provide comprehensive services including the use of HTS technology, compounds in various libraries, a database of results from screens and lead optimization. On a collaborative basis, HTS has the capability to support cellular and biochemical assays using absorbance, fluorescent kinetics, fluorescence resonance energy transfer, AlphaScreen, bioluminescence and cellular fluorescence imaging. In addition, HTS has expertise in adapting those biological and biochemical bench-top assays into high-throughput screening settings. Our HTS libraries are designed for diversity around not only well-established pharmacophore, but also very strict molecular property profiles that were balanced between diversity, physicochemical favorability, intrinsic complexity, and synthetic tractability. Encouraged by the fact that a significant number of marketed drugs are derived from natural products, we are also interested in expanding natural chemical products in compound libraries and developing an effective strategy to diversify the core backbone structure of natural products. We will design and explore biomimetic synthetic pathways to build up the molecule efficiently. Easily modified structure and high selective strategy will be preferred. The organocascade type assembling of complex structures will eventually result in the discovery of novel bio-valuable molecules to diversify the library. Biomedical and clinical property profiles will be highly considered for the library design and construction process.